A research article recently published in the journal Molecular Biology and Evolution provides an investigation into phylogenetic methods used to investigate the evolution and spread of COVID-19. The paper called "Phylogenetic Analysis of SARS-CoV-2 Data Is Difficult" analyses SARS-CoV-2 evolution using a data-driven approach and provides a review of the difficulties of inferring reliable phylogenies of the SARS-CoV-2 virus.
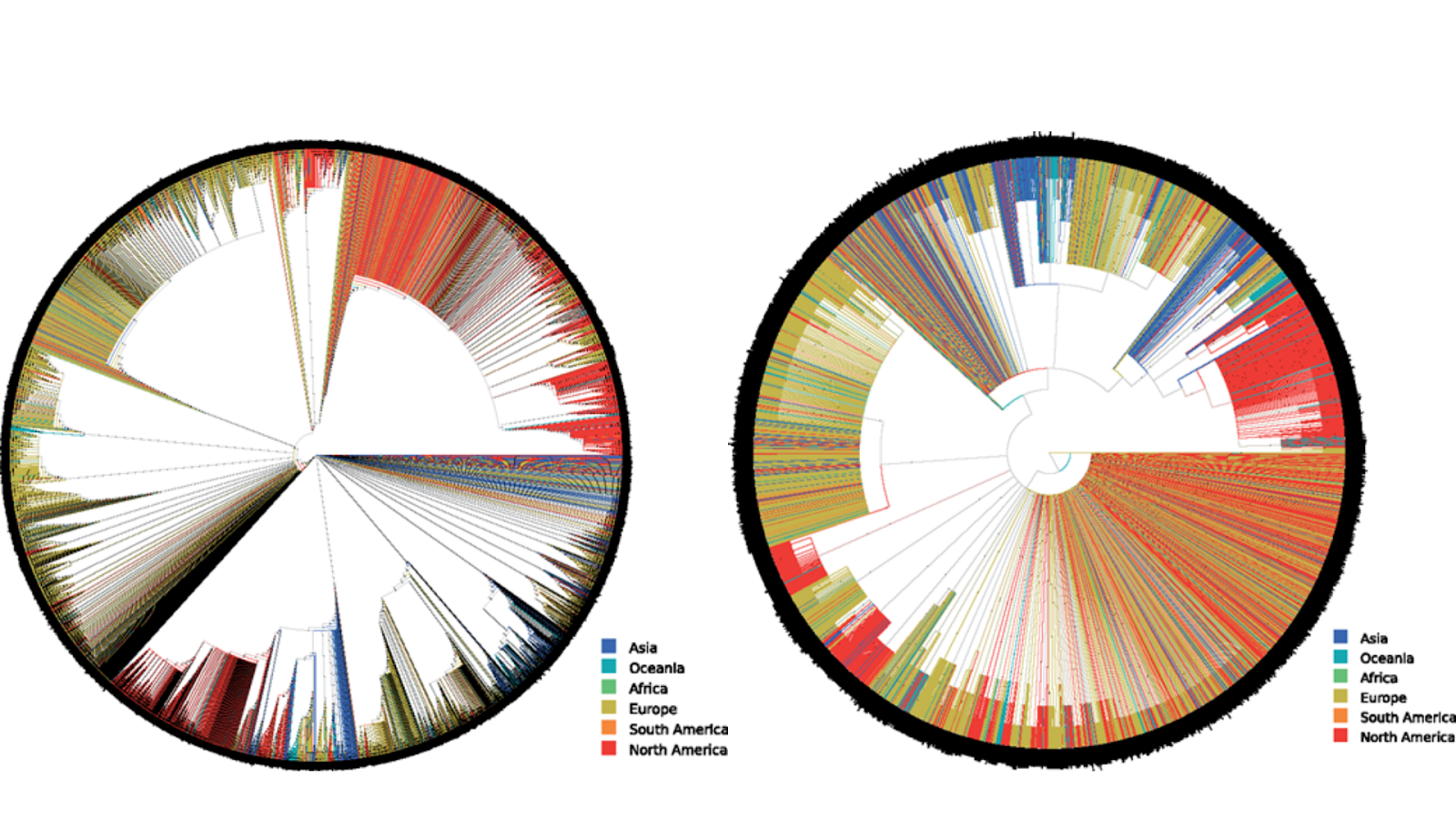
Extended majority rule consensus tree (FMSAO-CE) of the plausible tree set of the FMSAO alignment (on the left), and Rooted SMSA Maximum Likelihood tree (on the right). Source: Benoit Morel, Pierre Barbera, Lucas Czech, Ben Bettisworth, Lukas Hübner, Sarah Lutteropp, Dora Serdari, Evangelia-Georgia Kostaki, Ioannis Mamais, Alexey M Kozlov, Pavlos Pavlidis, Dimitrios Paraskevis, Alexandros Stamatakis, Phylogenetic Analysis of SARS-CoV-2 Data Is Difficult, Molecular Biology and Evolution, 2020;, msaa314, https://doi.org/10.1093/molbev/msaa314
The authors of the article, amongst whom the IGNITE researchers Prof. Alexandros Stamatakis and Ben Bettisworth, investigate the phylogenies of the SARS-CoV-2 virus using computational modelling techniques for genetic analysis, among which the RootDigger rooting tool developed within IGNITE. The team performs multiple sequence alignment, phylogenetic inference, and gene tree rooting to model the SARS-CoV-2 gene trees based on geographical regions, as well as region of origin.
Read the full article here.